Liver Cancer: pre-Malignant Stiffening, Membrane Transduction, & Nuclear Rheology
Project 1: Mechanisms of stiffening in liver cancer
Project 2: Can Soft Signals Turn Oncogenic?
In project 2 of Penn PSOC, we test the overall hypothesis (Figure 1), “Can Soft Signals Turn Oncogenic?” Can persistent “soft” signals (i.e. triggered through stiffness changes in the extracellular environment and changes in tension/ curvature distribution on the cell membrane) rewire the cell signaling to reflect the hallmarks of cancer in hepatocellular carcinoma (HCC)? The project is a collaboration between physical scientists Ravi Radhakrishnan, Tobias Baumgart, John Crocker, and Cell/Cancer Biologists Wei Guo and Mark Lemmon. The project investigators closely coordinate their studies with the clinical core investigators and the investigators of the theory core.
The aims of the project include: (1) Effect of substrate stiffness on receptor tyrosine kinase (RTK) and small GTPase signaling where the focus is on investigating biased signaling by growth factor receptor tyrosine kinases. (2) Effect of microenvironment on intracellular trafficking where the focus is on Microenvironment stiffness-induced exosome secretion and oncogenic signaling, and immune modulation. (2) We also investigate how are the physical properties (Figure 2) of the cell membrane signaling environment (membrane excess area, membrane tension, membrane and cytoskeletal interactions) in hepatocytes and stromal cells altered by the physical microenvironment variables relevant to HCC. Here, the focus is on the development of quantitative assays for the measurement of physical variables such a membrane tension and cortex pinning using optical tweezers as well as particle tracking. The experimental investigations are complimented by soft matter theory and multiscale simulations of membrane elasticity, membrane-cytoskeletal interactions, and signaling.
Clinical relevance: The outcomes of the studies in this project will directly lead to: (1) mechanotyping of live cells from patient derived tissues and liver matrices, (2) non-invasive monitoring of disease progression, and response to therapy including immunotherapy by analysis of patient-derived exosomes, and (3) design principles for targeted therapy in HCC through polypharmacology approaches to block oncogenic signaling.
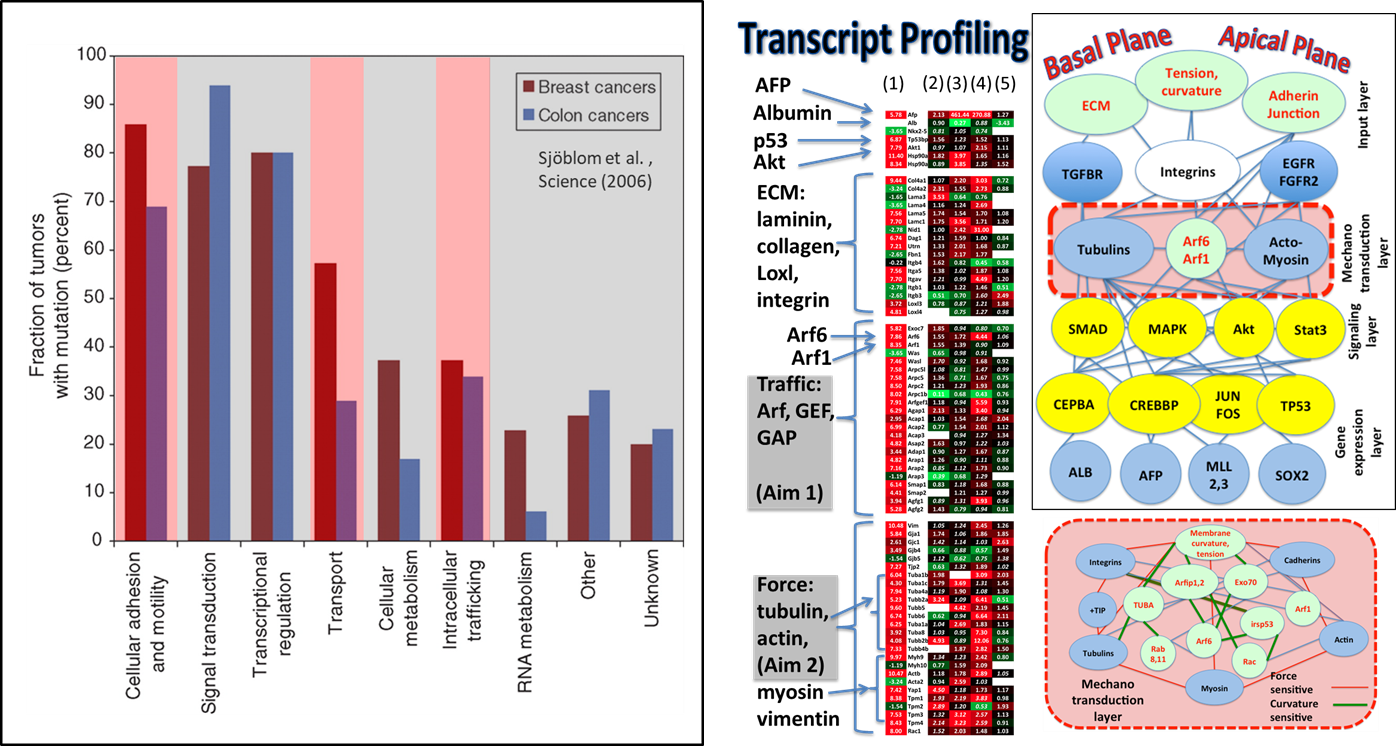 Figure 1: Overall Hypothesis: There exists causal mechanistic relationship between the physical characteristics of the hepatocellular carcinoma (HCC) physical microenvironment (cell membrane tension, compressive stress, substrate stiffness, matrix stiffness, growth factor exposure), and membrane-mediated signaling (trafficking of cell surface receptors; GTPase-mediated signaling).
Figure 1: Overall Hypothesis: There exists causal mechanistic relationship between the physical characteristics of the hepatocellular carcinoma (HCC) physical microenvironment (cell membrane tension, compressive stress, substrate stiffness, matrix stiffness, growth factor exposure), and membrane-mediated signaling (trafficking of cell surface receptors; GTPase-mediated signaling).
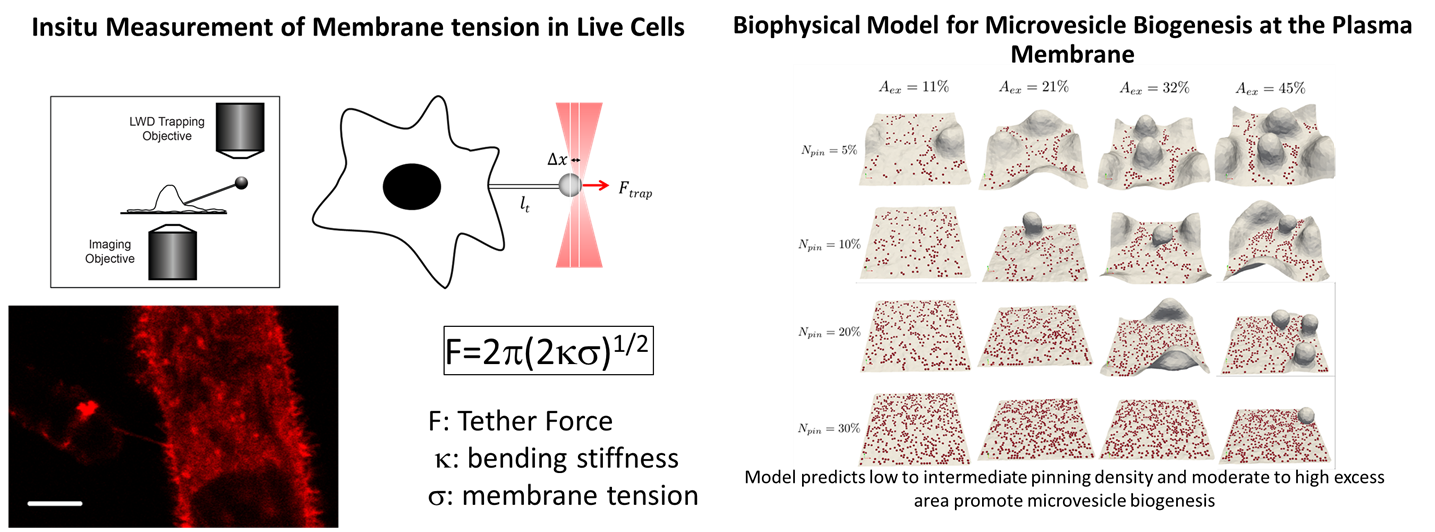 Figure 2: Biophysical assays and modeling methods for investigating the effect of physical variables of the microenvironment at the subcellular resolution.
Figure 2: Biophysical assays and modeling methods for investigating the effect of physical variables of the microenvironment at the subcellular resolution.
Project 3: Nuclear Rheology & Stability in Cancer
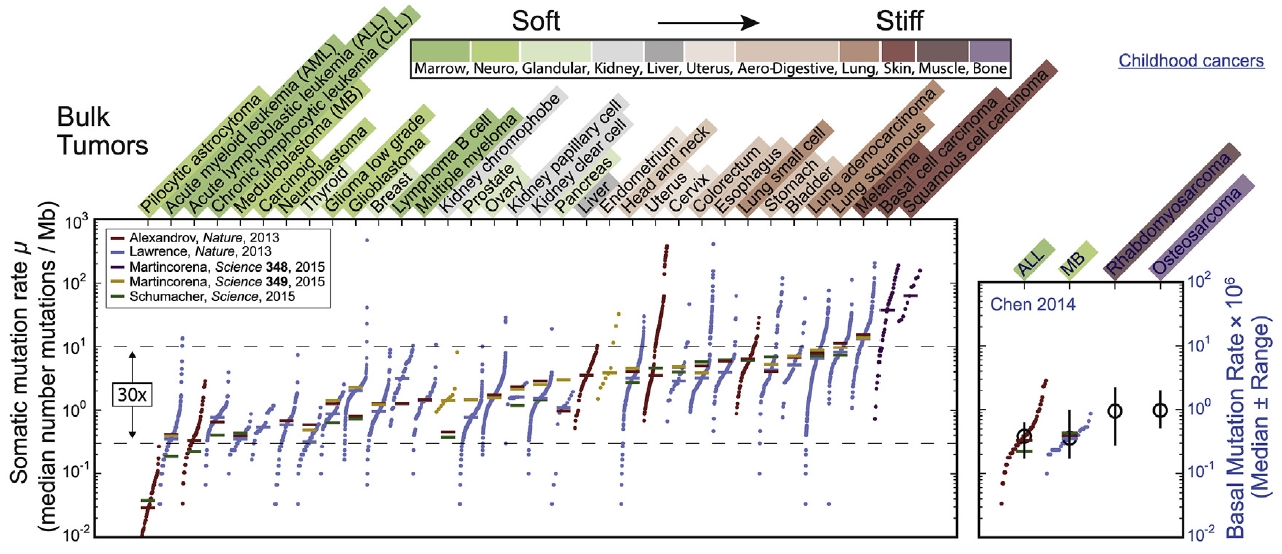
Many different types of soft and solid tumors have now been sequenced, and meta-analyses suggest that genomic variation across tumors scales with the stiffness of the tumors’ tissues of origin. Vogelstein and Tomasetti recently suggested cancer risk scales sub-linearly with the normal rate of stem cell divisions in the tissue of origin, suggesting a key role for DNA replication in mutation processes, and multiple ‘mechanogenomics’ mechanisms might explain the additional scaling of mutation rate with tissue stiffness. Since stiff solid tissues have higher density of fibrous collagen matrix, which should decrease tissue porosity, processes in proliferation could be affected and so could invasion into stiff tissues as the nucleus is squeezed sufficiently to enhance DNA damage. Although careful analyses continue to be required for rigorous conclusions about such DNA damage, diversification of a cancer genome after constricted migration is now clear in vitro. Understanding genome changes that give rise to neo-antigens is important to selection of cancer cell subpopulations (immunoediting) as well as to the development of immunotherapies. Monocytes/macrophages that might now be engineered to effectively attack cancer cells seem particularly relevant to understanding infiltration into solid tumors and the effects of microenvironment stiffness on genes such as the nuclear lamins. (Pfiefer CR et al. 2017)
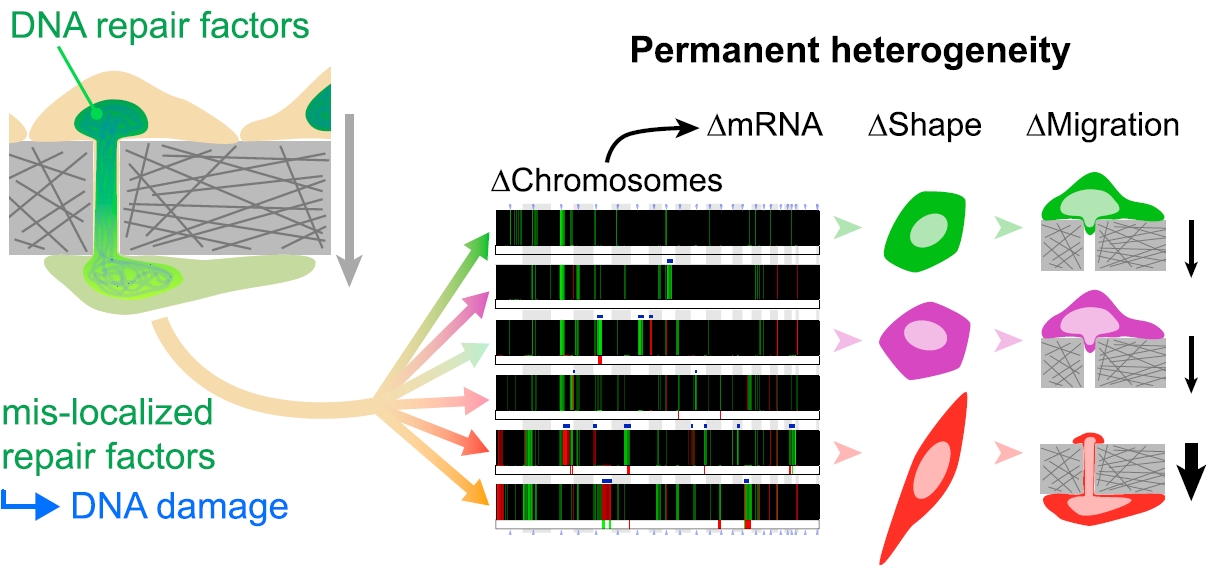
Migration through micron-size constrictions has been seen to rupture the nucleus, release nuclear localized GFP, and cause localized accumulations of ectopic 53BP1—a DNA repair protein. Here, constricted migration of two human cancer cell types and primary mesenchymal stem cells (MSCs) increases DNA breaks throughout the nucleoplasm as assessed by endogenous damage markers and by electrophoretic ‘‘comet’’ measurements. Migration also causes multiple DNA repair proteins to segregate away from DNA, with cytoplasmic mis-localization sustained for many hours as is relevant to delayed repair. Partial knockdown of repair factors that also regulate chromosome copy numbers is seen to increase DNA breaks in U2OS osteosarcoma cells without affecting migration and with nucleoplasmic patterns of damage similar to constricted migration. Such depletion also causes aberrant levels of DNA. Migration-induced nuclear damage is nonetheless reversible for wild-type and sub-cloned U2OS cells, except for lasting genomic differences between stable clones as revealed by DNA arrays and sequencing. Gains and losses of hundreds of megabases in many chromosomes are typical of the changes and heterogeneity in bone cancer. Phenotypic differences that arise from constricted migration of U2OS clones are further illustrated by a clone with a highly elongated and stable MSC-like shape that depends on microtubule assembly downstream of the transcription factor GATA4. Such changes are consistent with reversion to a more stem-like state upstream of cancerous osteoblastic cells. Migration-induced genomic instability can thus associate with heritable changes. (Irianto J et al. 2017)
Core 1: Cell and Tissue Core
Core 2: Theory Core – Multiscale models for mechano-chemical phenomena